Genetiska laboratorieanalyser
|
||||
| Genetiska laboratorieanalyser har sedan de introducerades kring år 1960 blivit en viktig del av den moderna sjukvården. Genetiska tester får allt fler kliniska användningsområden tack vare att kunskapen om genetiska orsaker till sjukdom har ökat i takt med de genteknologiska framstegen. Denna översikt syftar till att sammanfatta principerna för genetisk testning, som ett stöd främst för sjukvårdspersonal som kommer i kontakt med sådana undersökningar. | ||||
| 1. Medfödda och förvärvade mutationer | ||||
|
Genetiska tester syftar till att identifiera mutationer, förändringar i vår arvsmassa, som kan orsaka sjukdom. Konsekvenserna av mutationer beror på flera olika faktorer. Viktigast är om en mutation är medfödd (konstitutionell, engelska germline) och därmed finns i alla kroppens (kärnförande) celler, eller om den är förvärvad och då är begränsad till den vävnad där mutationen uppstått. Medfödda mutationer kan överföras till barnen medan förvärvade mutationer inte går i arv. En förvärvad mutation kan dock ge upphov till allvarlig sjukdom om den påverkar celldelningen så att en cancer uppstår. För att upptäcka om ett barn har en medfödd kromosomförändring görs kromosomanalys av lymfocyter från perifert blod. Om man vill identifiera kromosomförändringar som är typiska för vissa leukemier testar man prov från benmärg. | ||||
| 2. Analys av vår arvsmassa | ||||
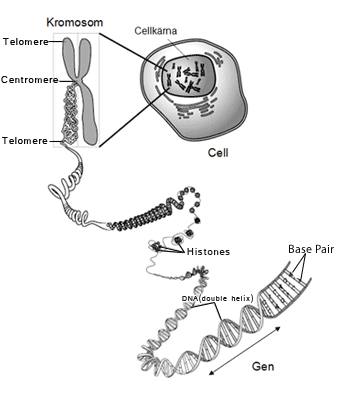 Figur 1:Kromosomer och DNA. Vid en rutinmässig kromosomanalys ser man cirka 450 enskilda kromosomband och bara genetiska förändringar som är så stora att de påverkar detta mönster kan upptäckas med cytogenetiska metoder. Under gynnsamma omständigheter kan en vanlig kromosomanalys upptäcka avvikelser som är mindre än 5 miljoner baspar men en rutinanalys förbiser ibland förändringar på 10-15 miljoner baspar. Cytogenetiska metoder lämpar sig därför som screeningmetod för stora avvikelser, till exempel när man misstänker att en patient har en hel kromosom extra (så kallad trisomi), eller när det fattas en stor del av en kromosom. Kromosomavvikelser behandlas i ett särskilt översiktskapitel i GenSvar.se. För att detektera mutationer i enskilda gener krävs analys med molekylärgenetiska metoder som PCR och DNA sekvensering. Sammantaget ger metoderna som illustreras i figur 2, möjlighet att detektera mutationer av olika storlekar, allt ifrån utbyte av en enskild bas till en hel förändrad eller extra kromosom. Därmed finns det idag tillförlitliga metoder som förmår att identifiera hela det spektrum av konstitutionella genetiska förändringar som orsakar medfödda sjukdomar (figur 2). 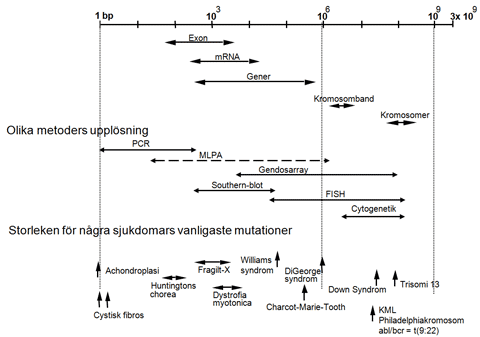 Figur 2: Bilden illustrerar några viktiga storlekssamband inom genetiken. Det totala haploida genomet omfattar 3 miljarder baspar och enskilda kromosomer mellan 50 och 300 miljoner baspar. De flesta generna innehåller omkring 10 000 baspar, men variationen är enormt stor. Många geners transkript (mRNA) är omkring 1000 baser långa uppdelade i medeltal 9 exon vilka i regel är några hundratal baser vardera. Som framgår i mitten på bilden kan mutationer som orsakar sjukdom vara av mycket varierande storlek, även för till synes ganska närbesläktade tillstånd. Sjukdomsframkallande mutationer kan vara alltifrån en enskild bas som bytts ut till att stora kromosomavsnitt saknas, finns i extra kopior eller har bytt plats. Symtom av en mutation avgörs inte av deras storlek utan av vilka konsekvenser den ger upphov till på proteinnivå. Således kan ett basparsutbyte helt förändra ett proteins struktur och leda till förändringar som är letala, medan en extra kopia av flera gener orsakad av en kromosomduplikation ibland kan ge ganska milda symtom. Den arsenal av metoder som står till buds baseras på flera principiellt olika metoder. Vid cytogenetik används vävnadsodling för att stoppa cellerna i celldelningens metafas och sedan studera kromosomerna. Med FISH (fluoroscent in situ hybridisering) används molekylära metoder för att analysera celler på objektsglas efter att dessa hybridiserats till fluoroscentmärkta sonder. Man använder antingen metafaskromosomer eller icke delande celler (interfas) för att visualisera kromosomavsnitt som är mindre än vad kromosomernas bandmönster kan upptäcka. Upplösningen för FISH är c:a 50 000 baspar och upp till en hel kromosom (chromosome painting). Notera att cytogenetik och vissa FISH-analyser fordrar att levande celler odlas vilket ställer särskilda krav på provmaterialet. Analyser av mindre genetiska förändringar i enstaka gener använder i stället DNA som kan isoleras ur till exempel vita blodkroppar, hudceller och även frusen vävnad. Isolerat DNA används även för att fastställa antalet genkopior som en screening av hela genomet med array-baserade analyser. Upplösningen är då beroende av antalet genprober som finns på den array som används. Southern-blot, en molekylär metod där DNA fragmenterats med restriktionsenzymer och storleksseparerats med elektrofores för att sedan hybridiseras till radioaktivt märkta sonder används för att detektera förändringar på 200 -300 baspar och upp till >50 000 baspar. Eftersom Southern-blot är en ganska komplicerad metod att utföra söker man ofta alternativ och metoden har därför minskat i betydelse på senare år. Med PCR kan man amplifiera genomiskt DNA i fragment som är upp till c:a 2000 baspar. Sedan kan dessa fragment analyseras på olika sätt för att identifiera mutationer. PCR är en enkel, robust och snabb metod, och det finns ett stort antal analyser för mutationsdetektion som baseras på denna metod. Ett exempel är MLPA (multiplex ligation probe amplification) som är en metod som möjliggör detektion av deletioner och duplikationer av olika storlek, från enstaka exoner till större kromosomregioner, vilket täcker mutationer i ett brett storleksintervall. De metoder som är inkluderade i figuren beskrivs mer utförligt nedan i detta kapitel. Notera att när man analyserar konstitutionella mutationer är man inte beroende av vilken vävnad som används som provmaterial. De genetiska förändringar man letar efter finns i kroppens alla celler, och man kan hitta mutationer i till exempel blodceller trots att symtom uppstår i en annan vävnad långt senare i livet. Det är alltså möjligt att i ett chorionvilli-prov (moderkaksprov) fastställa om ett foster riskerar att utveckla en muskelsjukdom i vuxen ålder. Vid genetiska analyser är det vanligast att man analyserar blodprover, men fosterceller (amniocyter eller chorionvilli-prov), vävnadsodlade hudfibroblaster, EBV-transformerade B-lymfocyter och saliv som innehåller celler från munslemhinnan används också. I vissa situationer kan sparade prover användas, till exempel intorkat blod från neonatal screening (så kallade ”PKU-lappar”), blod eller benmärgsutstryk som sparats på objektsglas, frusna tumörbiopsier eller formalinfixerade paraffininbäddade vävnadsklossar. Paraffinklossar utgör en stor biobank där det ofta återfinns material från avlidna personer, vilket kan vara av stort värde i släktutredningar. Tyvärr fungerar detta material inte alltid så bra för genetiska tester eftersom formalinbehandlingen kan ha degraderat DNAt så att den tekniska kvaliteten blir dålig. De epigenetiska förändringar som påverkar geners uttryck i olika vävnader genom metylering av baser i DNA eller modifiering av histoner är också svåra att studera. Metyleringsmönstret i konstitutionell vävnad analyseras rutinmässigt för att identifiera avvikande genomisk prägling (engelska imprinting) av vissa gener och kromosomregioner. Metoder för att analysera andra epigenetiska förändringar som är av betydelse vid utveckling och cancer har ännu inte satts i kliniskt bruk. | ||||
| 3. Monogena och Multifaktoriella sjukdomar | ||||
|
Möjligheterna till genetisk testning är idag framförallt beroende på vilken kunskap vi har om den aktuella sjukdomen, och hur sambandet mellan genetisk förändring och sjukdom ser ut. Det finns en uppsjö av bra metoder som klarar av att identifiera sjukdomsframkallande konstitutionella mutationer. Frågan är inte hur man skall testa, utan när och varför. Svårigheten med genetisk diagnostik är därför mindre teknisk än medicinsk. En monogen sjukdom orsakas av mutationer i en specifik gen, och sjukdomen nedärvs enligt Mendels lagar. För dessa sjukdomar föreligger ett mycket starkt samband mellan genotyp och fenotyp, det vill säga samband mellan genetisk förändring och symptom. Om en person bär det typiska basparsutbytet i kodon 380 i FGFR3 -genen kommer detta alltid att leda till en förändrad broskutveckling i de långa rörbenens tillväxtzoner som resulterar i dvärgväxt (achondroplasi). En mutation i en gen-kopia (en allel) ger en enhetlig klinisk bild. För de flesta tillstånd som ärvs enligt Mendels lagar, och där man identifierat sjukdomsmekanismen på molekylär nivå ses tydliga samband mellan mutation och symptom.
För så kallade multifaktoriella tillstånd är situationen betydligt svårare. Många av de vanliga folksjukdomarna som diabetes, hjärt-kärlsjukdomar, allergiska tillstånd, och demens tillhör denna kategori. Dessa sjukdomar anrikas ofta inom familjer, men något tydligt nedärvningsmönster kan ej urskiljas. Detta orsakas av att flera olika genetiska komponenter samverkar med livsstil och miljö för att symptom ska uppkomma. För flera multifaktoriella sjukdomar har genetiska faktorer kunnat identifieras på molekylär nivå. Ofta är dessa vanliga sekvensavvikelser, normalvarianter (polymorfier) i viktiga gener vilka är vanligare hos patienter med en viss sjukdom jämfört med friska individer. En viss normalvariant (4) av proteinet APOE förekommer hos 30% i befolkningen och är associerad med demens. Om en man inte har någon kopia av riskallelen 4 är risken för demens c:a 5 %, medan en kopia av riskallelen ökar risken till 12% och för män med två kopior (homozygot 4/4) är risken för demens cirka 35%. Det kan finnas flera andra genetiska polymorfier, i den aktuella genen eller i andra gener, som ökar eller minskar risken, och dessutom kommer livsstilen att påverka risken. Det är möjligt att identifiera genetiska förändringar hos patienter med folksjukdomar och deras anhöriga, men det är betydligt svårare att tolka och använda resultatet av ett test. Hur mycket ökar risken? Vilka andra genetiska och omgivningsfaktorer påverkar denna risk? Kunskapsutvecklingen kommer att leda till att man identifierar många olika genetiska riskfaktorer som tillsammans kan ge en ännu bättre prognos. Innan man utför tester måste man dock först ta ställning till testresultatets kliniska betydelse för den enskilda patienten, det vill säga om resultatet leder till annan behandling eller preventiva åtgärder. Till skillnad från testning för monogena sjukdomar ger genetisk testning vid komplexa sjukdomar inte tillräckligt specifik information för att påverka omhändertagandet av en enskild patient eller släkting. | ||||
| 4. Mutationsscreening | ||||
|
Genetiska laboratorietester utförs ofta för att bekräfta diagnosen av en misstänkt monogen sjukdom hos en sjuk patient. En monogen sjukdom orsakas av mutationer i en specifik gen, men oftast förekommer det inom en population många olika sjukdomsframkallande mutationer i genen, det vill säga alla patienter med den specifika sjukdomen har mutation i samma gen, men mutationen kan se olika ut och vara belägen på olika ställen i genen. Detta medför att undersökningen av den första patienten i en släkt ofta är en ganska komplicerad och omfattande screening.
Om man kan påvisa en mutation hos en patient innebär det att: 1) Man får en diagnosmisstanke bekräftad. 2) Mutationen kan ge en viss indikation på sjukdomens förväntade förlopp eftersom det ofta föreligger ett genotyp- fenotyp samband. 3) Andra släktingar kan erbjudas anlagstestning. 4) Vid en graviditet kan fosterdiagnostik eventuellt erbjudas. Screening för mutationer som en del i en klinisk utredning av en misstänkt genetisk sjukdom kan dock resultera i att man inte hittar någon mutation. Skälen till detta är flera. Det kliniska underlaget för att det rör sig om en ärftlig sjukdom kan vara svagt, men man vill utesluta ett allvarligt ärftligt tillstånd. Andra gånger är genetiken komplex, och kanske kan sjukdomen även orsakas av mutationer i andra, ännu inte identifierade, sjukdomsgener. Frånvaron av mutation i en analyserad gen har då litet kliniskt värde. Om man däremot hittar en mutation är det diagnostiskt och därmed viktigt för patienten och patientens familj/släkt . | ||||
| 5. Anlagstestning | ||||
|
Om en ärftlig monogen sjukdom nedärvs i en familj kan en riktad mutationsanalys, anlagstestning, erbjudas till släktingar. Anlagstestning förutsätter att man har identifierat den mutation som segregerar i släkten, det vill säga att man först analyserat en säkert sjuk släkting och identifierat en mutation. Anlagstestet blir sedan en riktad undersökning som säkert kan fastställa om en släkting bär denna mutation eller inte. Den första screeningen i släkten är oftast omfattande och därmed dyr. En riktad anlagstest är i stället enklare eftersom man då vet vad man skall leta efter. Vid sent debuterande dominanta sjukdomar kan anlagstestning fastställa om en frisk person har hög risk att insjukna senare i livet. Vid recessiva sjukdomar kan nära släktingar ta reda på om de i sin tur riskerar att få sjuka barn. Riktad mutationsanalys kan även utföras vid fosterdiagnostik.
En andra förutsättning för en anlagstest är att de personer som genomgår anlagstestning har erbjudits genetisk vägledning. Även om själva testet är enkelt att utföra är de medicinska, psykologiska, sociala och etiska aspekterna på anlagstestning ofta komplexa. Det faktum att man rent tekniskt kan ta reda på om en person bär på ett sjukdomsanlag betyder inte att personen själv vill ha denna information. Resultatet kan få betydelse för personens fortsatta liv på ett sätt som han eller hon inte kan överblicka, utan att först erhålla information och sedan tänka efter i lugn och ro. | ||||
| 6. Genotypning och Kopplingsanalys | ||||
I familjer med en monogen sjukdom lyckas man ofta identifiera den sjukdomsframkallande mutationen, och kan därefter erbjuda riktad mutationstest för anlagstestning. I vissa situationer kan man, även om mutationen inte har identifierats, använda en indirekt metod för att följa hur sjukdomsanlaget ärvs i en familj. Denna indirekta metod, kopplingsanalys, kan användas för anlagstest eller fosterdiagnostik. Metoden bygger på att man analyserar normala sekvensvariationer i DNA (polymorfier) som markörer för en gen. De vanligaste polymorfierna utgör variationer i repeterade sekvenser (t.ex. CACACACACA) belägna i intron eller intill gener och alltså inte påverka genernas funktion. Genom att analysera familjemedlemmar för en markör kan man följa hur olika marköralleler nedärvs i relation till hur sjukdomen nedärvs inom familjen. På så sätt kan en viss markörallel ”kopplas” till sjukdomsanlaget i den specifika familjen. Vid diagnostik väljs markörer som är belägna inom eller mycket nära sjukdomsgenen. En förutsättning för kopplingsanalys är att man har tillgång till material från flera familjemedlemmar, inklusive en sjuk individ. 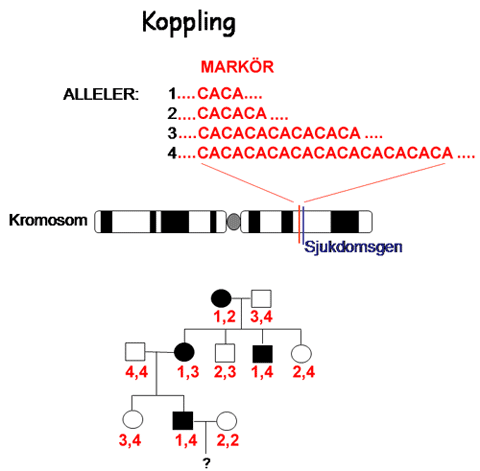
Figur 3: Ett locus (ett specifikt ställe i genomet) med varierande antal CA, en sk mikrosatellitmarkör. Varje locus kan lätt analyseras med PCR där man utnyttjar de unika sekvenser som flankerar CA-repetitionen för att amplifiera DNA. Storleken på de PCR amplifierade fragmenten kan bestämmas och storleken beror på antalet mellanliggande CA. I familjen kan man följa dessa alleler (varianter i locuset betecknade 1-4) i en familj. I det humana genomet finns många tusen liknande polymorfa repetitioner spridda längs alla kromosomer. 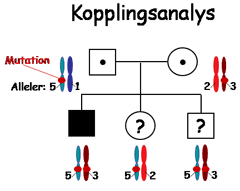
Figur 4: Illustreration av hur man med kopplingsanalys kan fastställa om ett foster bär anlaget för cystisk fibros (CF). Bilden visar att det första barnet har CF och mutationsscreening visade att barnet bär en kopia av den vanligaste CF-mutationen, ∆F508. CF är ett autosomalt recessivt tillstånd som fordrar att genen är muterad både på den kromosom som kommer från mamma och den som kommer från pappa. Man fann dock inte den andra mutationen vid mutationsscreeningen. I den aktuella familjen använder man sig i stället av kopplingsanalys för att genomföra fosterdiagnostik. Siffrorna (1-5) anger olika normalvarianter i en sekvenspolymorfi i sjukdomsgenen CFTR. Med enkel PCR kan man skilja på pappas bägge kromosomer, liksom mammas. Kopplingsanalysen visar att den sjuka dottern har ärvt en paternell 5-allel som sitter på samma kromosom som F508. Mamma har genotypen 2,3 dvs hon har en 2-allel på ena kromosomen och en 3-allel på den andra. Eftersom den sjuka dottern har ärft mammas 3-allel vet man att denna allel sitter på samma kromosom som den icke identifierade maternella CF-mutationen. Vid nästa graviditet kan man genotypa fostret med DNA som isolerats från en chorion-villibiopsi. Foster som ärver både faderns 5-allel och moderns 3-allel blir sjuka. Avsaknad av en eller båda av dessa sjukdomskopplade alleler resulterar i barn som inte utvecklar CF. Kopplingsanalys används framför allt vid fosterdiagnostik vid monogena sjukdomar som orsakas av en enda gen. En annan användning av kopplingsanalys är vid preimplantatorisk genetisk diagnostik (PGD), eftersom genotypning av polymorfier är en mer robust metod än en specifik mutationstest när man bara har en enda cell att tillgå. Eftersom man kan använda polymorfa markörer för att särskilja en individs kromosomer har genotypning av mikrosatellitmarkörer utvecklats som ett sätt att indirekt fastställa antalet kromosomer av en viss typ. Detta är principen för quantitative flourescent PCR (QF-PCR) som används vid fosterdiagnostik av trisomier. Genom att använda markörer belägna på kromosom 13, 18 och 21 kan man fastställa om fostret har två eller tre kopior av dessa kromosomer. Metoden är ett enklare och snabbare alternativ till vanlig kromosomanalys för att upptäcka trisomi 13, 18 och 21. Med QF-PCR gör man bara en genotypning och fastställer antalet alleller hos fostret, utan att analysera föräldrarna. Vid QF-PCR analys ingår idag markörer för kromosom 13,18, 21, X och Y. Genotypning används även inom rättsgenetik för faderskapsbestämning, för att identifiera kvarlevor av avlidna samt för att spåra brottslingar. | ||||
| 7. Metoder för mutationsanalys | ||||
|
GenSvar har ett särskilt kapitel om kromosomavvikelser där karyotypering och FISH behandlas. Principerna för fosterdiagnostik behandlas också i ett särskilt översiktskapitel. Nedan följer en sammanfattning av de metoder som används vid DNA-baserad diagnostik av ärftliga sjukdomar. För flera av dessa metoder finns olika alternativ men vi har valt att beskriva de metoder som är vanligast inom genetisk diagnostik. PCR Polymerase chain reaction (PCR) är en metod (uppfunnen av K Mullis), som möjliggör att man specifikt amplifierar ett DNA-fragment i miljoner kopior från mycket små mängder DNA. Metodens specificitet bygger på de primers, ca 20 baser långa syntetiska oligonukleotider, vars sekvens väljs ut med ledning av människans kända DNA-sekvens. Om man designar dessa primers på rätt sätt kommer de bara att kunna baspara (eng: annealing) sig till ett specifikt (komplementärt) ställei det humana genomet. För att amplifiera en önskad DNA sekvens designas två primers så att de är riktade mot varandra och flankerar den region man är intresserad av. Amplifieringen bygger på att man i närvaro av ett termostabilt DNA-polymeras och syntetiska nukleotider låter provet genomgå cykliska temperaturförändringar (figur 5). Det är därmed möjligt att kopiera enbart det mellanliggande segmentet så att man från 50 ng genomiskt DNA (som innehåller hela genomet) erhåller 1 mikrogram av det amplifierade fragmentet. Den sekvens man vill amplifiera utgör mindre än en miljondel av allt templat-DNA, men efter 20 PCR-cykler har man flera miljoner kopior av detta fragment. Denna PCR-produkt kan sedan användas för vidare analys på olika sätt, till exempel DNA sekvensering. 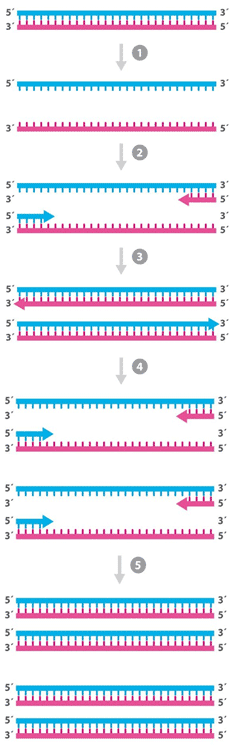
Figur 5: Det dubbelstängade DNA templatet denatureras genom att värma reaktionen till 95°C (1). PCR primers tillåts baspara (annealing) till sin specifika målsekvens vid optimal temperatur (50-65°C) (2). I närvaro av termostabilt DNA-polymeras, syntetiska nukleotider, och buffert kan en komplementär DNA sträng bildas genom inkorporering av nukleotider och elongering av DNA strängen vid 72°C (3). Från en kopia av DNA templatet har man nu två kopior, där varje DNA molekyl består av en gammal och en nysyntetiserad DNA sträng. Reaktionen kan nu genomgå nästa cykel med denaturering, annealing och elongering, vilket resulterar i fyra kopior av DNA. Vanligen får reaktionen genomgå 25-35 cykler. (Bildkälla: A Read & D Donnai, New Clinical Genetics, ©Scion Publishing Ltd)
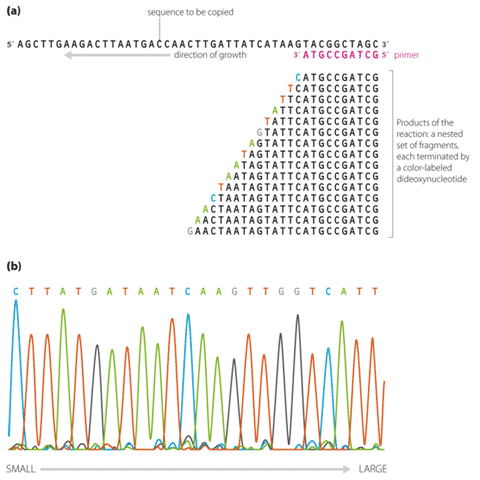
Figur 6: Man gör först en PCR-amplifiering av det eller de exon som skall sekvenseras. Stora exon delas upp i flera mindre överlappande PCR-produkter. Sekvensreaktionen innehåller PCR-produkt (templat), sekvensreagenser, en primer (Forward eller Reverse för att syntetisera en sträng i taget), DNA-polymeras, fyra olika nukleotider, samt fyra olika dideoxynukleotider. De fyra dideoxynukleotiderna (terminatorerna) är flouresecensmärkta med var sin fluorofor, (A=grön, T=röd, G=svart och C=blå). Koncentrationen av terminatorerna är låg, så varje gång en nukleotid inkorporeras i den växande DNA strängen, finns en liten chans att det är en dideoxynukleotid, vilket sätter STOPP för syntesen. Resultatet blir ett antal DNA fragment som skiljer sig i längd med en nukleotid där den sista nukleotiden är fluorescensmärkt. (Bildkälla: A Read & D Donnai, New Clinical Genetics, ©Scion Publishing Ltd) 
Figur 7: Efter sekvensreaktionen separeras syntesprodukterna på en DNA-sekvenator. Detta instrument har kapillärer fyllda med en gelmatrix genom vilket DNA separeras efter storlek (längd) med elektrofores. Till kapillärerna är en laserlampa och en CCD-kamera kopplade som gör att man kan detektera de fyra olika färgerna. Färgerna registreras elektroniskt och ordningen på färger (dvs nukleotider) sammanställs baserat på tiden det tog att passera detektorn. Denna tid är proportionell mot DNA-fragmentets längd och varje sådant fragment har en färg motsvarande sista inkorporerade nukleotiden (A= grönt etc). På så sätt får man fram ett sekvensdiagram där varje bas är representerad i tur och ordning. Vanligen kan man tillförlitligt läsa 300-600 baser i en sekvens. Denna sekvens exporteras därefter elektroniskt till ett dataprogram där kvaliteten på sekvensen kan utvärderas och jämföras med en känd sekvens. I klinisk rutin brukar man utföra en sekvensreaktion av varje sträng i DNA-molekylen (forward respektive reverse) och jämföra bägge strängarna mot den kända sekvensen. Vid pilen ses två olika baser, G och C, vilket betyder att i denna position har individen två genkopior med olika baser i denna position. (Bildkälla: A Read & D Donnai, New Clinical Genetics, ©Scion Publishing Ltd)
 Figur 8 Oligonucleotide ligation assay – OLA OLA lämpar sig för applikationer där man vill analysera ett begränsat antal mutationer samtidigt i en patient. Genom att designa prober bestående av parade oligonukleotider, specifika för normal respektive muterad sekvens, och som kan göras olika långa och med olika fluorescens kan man lätt testa för 30 olika mutationer i en reaktion. Denna assay används kliniskt bland annat vid analys av Cystisk fibros, då man analyserar de 33 vanligaste förekommande mutationerna.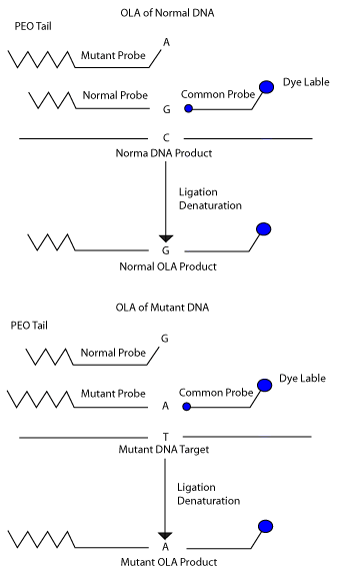
Figur 9: OLA bygger på att två oligonukleotider kan ligeras samman om de är homologa och hybridiserar till intilliggande sekvenser. Antag att man vill testa för en mutation där basen C bytts ut mot T i en gen. Proben består då av en oligo (common probe) som hybridiserar till sekvensen direkt efter den aktuella basen. Den andra oligon hybridiserar till intilliggande sekvens och med den aktuella basen i 3’ änden. Man kan då välja om den oligon ska vara specifik för normal sekvens eller muterad sekvens. Man kan också syntetisera två olika oligos med olika längd, en specifik för normal sekvens och en specifik för muterad sekvens. Vid en hybridisering av dessa oligos samt efterföljande ligering kommer bara den oligonukleotid som är exakt homolog till templatet att resultera i en ligeringsprodukt, det vill säga antingen oligon specifik för mutationen eller oligon specifik för normal sekvens har ligerats till den gemensamma oligon. Om en ligering sker kommer produkten att hänga ihop som ett fragment. Produkterna separeras sedan med elektrofores, och produkter som innehåller C respektive T i den kritiska positionen kan särskiljas antingen genom olika längd och/eller olika fluoroforer (färger) som bundits till oligonukleotiden. På så sätt kan man fastställa om det DNA man analyserar har C eller T i den position man analyserar. Vid Oligonucleotide ligation assay (OLA) kan man testa för upp till 30 olika mutationer i en och samma reaktion. Steg ett är multiplex PCR amplifiering av de olika aktuella regionerna. En specifik probe för varje mutation tillsätts sedan till PCR produkterna. Varje probe består av tre oligonukleotider som hybridiseras till PCR produkterna, följt av ligering och storleksbestämning av ligeringsprodukter. Restriktionsenzymanalys, RFLP Många mutationer påverkar DNA-sekvensen så att ett restriktionsenzym ändrar sitt klyvningsmönster jämfört med den normala sekvensen och ger då upphov till en mutationsspecifik RFLP (Restriction fragment length polymorphism). Om den normala sekvensen omfattas av igenkännings-sitet för ett restriktionsenzym kommer en mutation i dessa baser att resultera att restriktionsenzymet inte klyver vid denna position. I andra situationer kan en mutation innebära att ett nytt restriktionssite skapats. 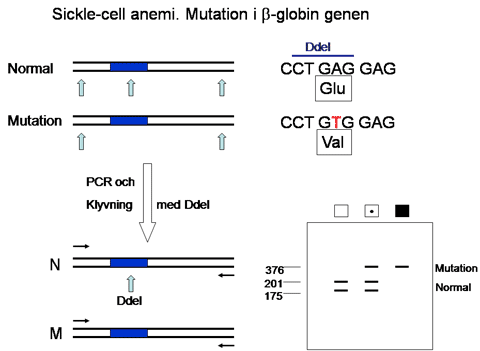 Figur 10: Bilden illustrerar hur man identifierar den vanligaste mutationen vid sickle-cell anemi. Först gör man en PCR-amplifiering av den eftersökta delen av genen och klyver med restriktionsenzymet. Klyvningsprodukterna separeras därefter med vanlig elektrofores på en agarosgel. Metoden har stor användning eftersom den är enkel och robust. Metoden är dock endast applicerbar om den aktuella mutationen är vanlig och utgör en betydande del av de förekommande mutationerna. Så är t.ex. fallet vid hemokromatos, akondroplasi och sickle-cell anemi. Southern-blot Southern-blot är en av de klassiska metoderna inom molekylärgenetik som på senare tid ersatts alltmer av PCR-baserade metoder. Southernblot är ännu den enda rutinmetod som finns för att identifiera stora trinukleotid-expansioner. Vid till exempel Fragilt-X eller Dystrofia myotonica kan expansionerna vara 1000 - 4000 baspar långa och kan därför inte amplifieras med PCR utan Southern-blot används i stället. 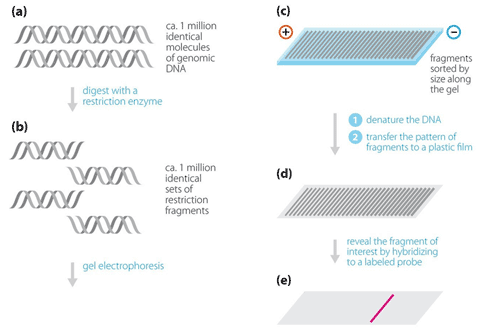 Figur 11: Vid Southern-blot klyvs DNA med restriktionsenzym. De fragment som på så sätt uppstår separeras på en agarosgel och överförs med kapillärkrafter till ett membranfilter (engelska blotting - jämför läskpapper) som binder DNA. Detta filter har alltså alla de DNA fragment som uppkom med klyvningen separerade efter storlek. Nästa steg är att hybridisera en radioaktivt märkt genspecifik sond till filtret. Efter att överskottsprobe tvättats bort lägger man filtret på en röntgenfilm (autoradiografi) varvid radioaktiva restriktionsfragment framträder som band. (Bildkälla: A Read & D Donnai, New Clinical Genetics, ©Scion Publishing Ltd) Gendosförändringar En gens funktion kan förändras av att enstaka baser påverkas, men även deletioner eller duplikationer av hela exon eller större bitar av en gen kan orsaka sjukdom. Ett antal återkommande syndrom orsakas av mikrodeletioner och mikroduplikationer, dvs deletioner eller duplikationer av kromosomregioner på omkring 1 miljon baspar eller mer, vilka kan innehålla ett flertal gener. Dessa mikrodeletioner/duplikationer uppkommer vanligt mellan repetitiva sekvenser i vissa kromosomregioner där rekombinationer kan resultera i att ett mellanliggande avsnitt går förlorat eller dupliceras. Liknande mekanismer kan resultera i att ett eller flera exon kan gå förlorat i en gen. Gendosförändringar (eng: Copy number variants, CNV) upptäcks inte med vanlig PCR eftersom metoden vanligen inte är kvantitativ. De ovan beskrivna metoderna är inriktade på att analysera sekvensmutationer och fungerar inte för att detektera deletioner, i stället detekteras gendosförändringar med FISH-baserade och ligeringsbaserade metoder eller sk CGH-array. Interfas-FISH Mikrodeletionssyndrom kan lätt analyseras med FISH på metafas kromosomer eller celler i interfas. Metoden bygger på att cellkärnor fixeras och torkas in på ett objektsglas. Detta glas hybridiseras därefter med en eller flera fluoroscensmärkta sonder (eng: probe) som vanligen är över 100.000 baspar långa och som innehåller den region som skall analyseras. Efter hybridisering och tvätt, för att avlägsna icke hybridiserad sond, analyseras glaset i ett fluorescensmikroskop. Varje hybridiserad sond ger upphov till en färgad signal (prick) som kan visualiseras (figur 12 och 13). Normalt har man en sådan signal per kromosom, men vid en deletion saknas en signal. Vid genamplifiering som är vanligt vid vissa tumörformer ses ett ökar antal signaler. Metoden är robust och fordrar ganska enkel utrustning men är svår att utföra i stor skala eftersom varje glas måste hybridiseras och analyseras för sig. FISH begränsas främst av att sonderna inte kan göras för små eftersom signalstyrkan då går förlorad. I klinisk rutin fungerar därför FISH för mikrodeletionssyndrom, och amplifieringar men kan i regel inte detektera gendos avvikelser som bara omfattar enstaka gener eller delar av gener. En viktig användning av interfas-FISH är också inom fosterdiagnostik och annan snabb-diagnostik av kromosomavvikelser. Med enkelkopieprober från kromosom 13, 18 och 21 kan man identifiera de vanligaste numeriska kromosomavvikelserna utan att behöva odla fosterceller eller celler från en patient. Därmed kan svarstiderna för fosterdiagnostik reduceras från 2 veckor till 1 dag och för diagnostik av till exempel trisomi 21 hos en nyfödd från 3 dagar till mindre än en arbetsdag. Interfas-FISH kan dock bara identifiera ett fåtal avvikelser i taget eftersom man inte kan använda mer än några få sonder per analys. 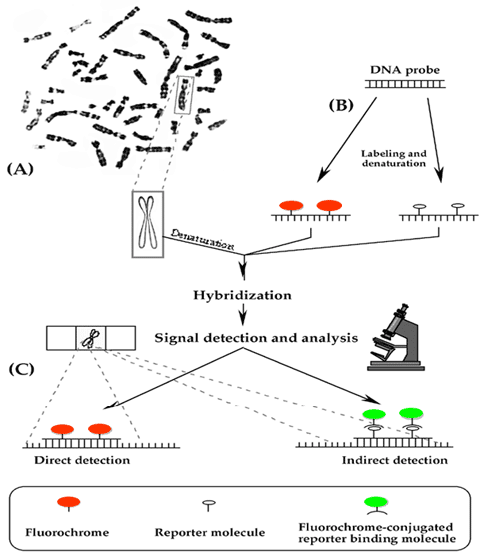 Figur 12. FISH-analys av metafaskromosomer. DNA sonder (Engelska: prober) kan märkas in direkt med fluorescent molekyl eller indirekt med en reportermolekyl (t ex biotin). Efter denaturering av metafaskromosomer samt denaturering av DNA sond kan dessa tillåtas hybridisera till varandra. DNA sonden binder till sin komplementära sekvens på kromosomen och efter tvätt, för att avlägsna överflödig sond, kan signaler detekteras via ett fluorescensmikroskop. Om sonden är indirekt inmärkt med reporter sker märkningen av fluorescens i efterhand via en molekyl som binder till reporter molekylen (t ex avidin). 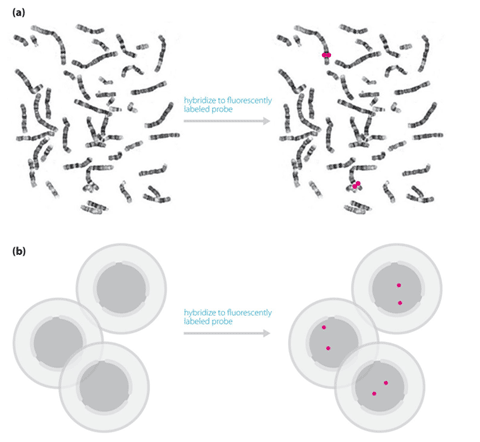 Figur 13. FISH analys kan utföras på metafaskromosomer (a) såväl som på cellkärnor som befinner sig i interfas (b). I det senare fallet ser man inte de enskilda kromosomerna, men antalet signaler i cellkärnan kan ge information om antalet kopior av den aktuella regionen man undersökt. (Bildkälla: A Read & D Donnai, New Clinical Genetics, ©Scion Publishing Ltd) MLPA FISH-tekniken är väl lämpad att identifiera gendosförändringar som omfattar större kromosomavsnitt. Det har visat sig att det också finns ett betydande behov att identifiera deletioner eller duplikationer av enstaka gener eller exon. För att kunna göra detta använder man en ligeringsbaserad metod, MLPA (multiplex ligation-dependent probe amplification), som illustreras i figur 14. För den sekvens man vill analysera designas två intilliggande oligonukleotider. Dessa hybridiseras till genomiskt DNA, varefter man ligerar dem, och sedan amplifierar dem med PCR. Endast oligos som är perfekt homologa till målsekvensen kan ligeras ihop, och därmed amplifieras med PCR. Antalet PCR produkter är då direkt proportionell mot antalet templatkopior. Oligonukleotider av olika längd med en universell primersekvens i ändarna kan användas samtidigt vilket gör att man i en och samma reaktion kan kombinera upp till 40 olika tester (om man använder fler färger kan få ännu fler) och därmed testa flera kromosomavsnitt eller flera exon i en gen. Eftersom man använder DNA i lösning och enhetliga primers för PCR lämpar sig MLPA för storskalig användning, det vill säga man kan testa många patienter samtidigt. 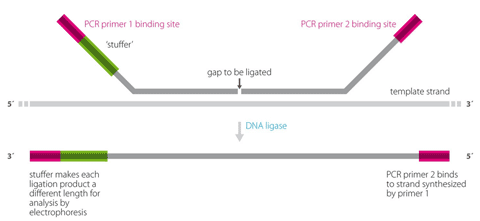 Figur 14: MLPA inleds med att man, för varje målsekvens man vill testa, hybridiserar två specifika oligonukleotider till genomiskt DNA. Dessa två oligonukleotider binder till intilliggande sekvenser, och endast vid hel komplementaritet kan de ligeras ihop. De oligonukleotider man använder har dessutom i ytter-ändan en PCR primersekvens samt däremellan olika långa mellanliggande avsnitt (stuffer sekvens). Nästa steg är att man gör en PCR-reaktion med hjälp av fluorescensmärkta universella primers, då endast ligeringsprodukter kommer att amplifieras. Antalet PCR-produkter kommer att vara proportionellt till antalet ligeringsprodukter som i sin tur beror på antalet templatkopior. Eftersom samma primers kan PCR-amplifiera alla ligeringsprodukter, och man använder oligonukleotider med olika långa mellanliggande avsnitt (stuffer sekvens), är det möjligt att utföra flera ligeringsreaktioner samtidigt i samma rör (multiplexing). (Bildkälla: A Read & D Donnai, New Clinical Genetics, ©Scion Publishing Ltd)  Figur 15: Genom sådan ”multiplexing” kan man i samma reaktion analysera 40 (se kommentar ovan) olika loci (gener/exoner/sekvenser), där varje lokus ger en PCR produkt med specifik längd. PCR-produkterna separeras på en sekvenator och mängden PCR produkt för varje lokus kvantifieras och jämförs med resultat från friska kontroller. För varje ligeringsprodukt (probe) beräknas ett ratio jämfört med frisk kontroll och om ingen deletion eller duplikation föreligger hos patienten för denna specifika målsekvensen ligger den normala ration omkring 1. CGH-array Comparative genomic hybridization (CGH) är en metod som utvecklades i början av 1990-talet framför allt för genetiska studier av cancerceller. Metoden bygger på att man isolerar DNA från en vävnad man vill testa (test-DNA) och märker in detta material med en fluorofor (till exempel röd fluoroscens). Ett referens-DNA som bäst isoleras från normal vävnad från samma individ märks med en annan färg (grön). Därefter blandas lika delar av dessa och hybridiseras till normala metafaskromosomer från en kontrollindivid. Därmed färgas varje kromosom med summan av grön och röd. Om en viss kromosom i cancercellerna är amplifierad kommer den röda färgen att överväga och om det fattas material i cancercellerna blir motsvarande kromosom mer grön. Färgförhållandet mellan grön och röd kan kvantifieras elektroniskt och på så sätt kan man identifiera om det föreligger en gendosavvikelse i tumörDNA. Upplösningen med CGH är sämre än med kromosomanalys, men eftersom det ofta är svårt att odla cancerceller har metoden ett värde som forskningsinstrument inom cancercytogenetik. Array-CGH bygger på samma princip som beskrevs ovan men i stället för att hybridisera blandningen av test-DNA och kontroll-DNA mot en metafas hybridiserar man mot en så kallad array med klonat DNA (figur 16). Metoden är betydligt känsligare än vanlig kromosomanalys för att upptäcka avsaknad eller extra kopior av ett kromosomsegment. Upplösningen beror på hur många kloner arrayen innehåller samt hur stor del av genomet dessa kloner representerar. Array-CGH används när man har misstanke på att en patient har en kromosomavvikelse som inte kan upptäckas med vanlig kromosomanalys (figur 17). Eftersom metoden förmår att identifiera såväl små förändringar som avsaknad eller extra kopior av en hel kromosom kan man ibland göra array-CGH som första analys vid utredning av misstänkt kromosomavvikelse. Om man inte kan identifiera någon förändring på en array-CGH som täcker hela genomet med tillräckligt antal sonder har man uteslutit obalanserade kromosomavvikelser och mikrodeletionssyndrom. 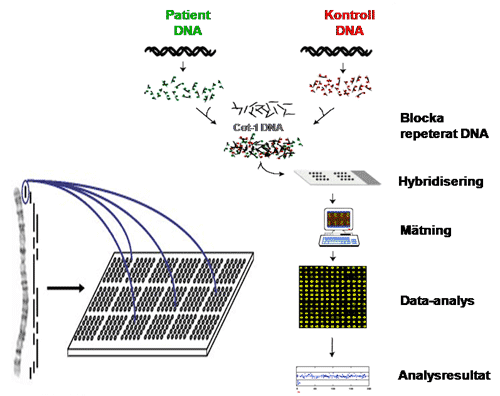 Figur 16: Genomiskt DNA isoleras från patient respektive normal kontroll och inmärks med olika fluoroforer (patient DNA med grön fluorofor och normalt DNA med röd fluorofor). Dessa DNA-prov blandas i lika stor mängd tillsammans med Cot-1 DNA (som blockerar repetitiva sekvenser), och tillåts hybridisera till en sk array. Vid array-CGH används vanligen BAC-kloner med human insert bestående av c:a 200.000 baspar genomiskt DNA. Dessa BAC-kloner kan ”spottas”, det vill säga sättas som små fläckar på ett objektsglas som sedan kan användas för att hybridisera mot. Färgförhållandet mellan grön och röd i varje ”spot” kan kvantifieras elektroniskt och på så sätt kan man identifiera om det föreligger en gendosavvikelse i patient DNA. Genom att välja BAC kloner med insert som överlappar litet får man sk ”tiling arrays” (jämför tegeltak). En tiling array med c:a 33.000 BAC-kloner innehåller således över 6 miljarder baspar och täcker därmed hela det humana genomet med i genomsnitt 2 kloner för varje del av genomet. Med CGH mot en tiling array kan man få en upplösning på ungefär 100.000 baspar, vilket skall jämföras med 10 miljoner baspar som är upplösningen vid rutinmässig kromosomanalys. I stället för BAC-kloner kan även oligonukleotider används, varvid arrayen innehåller fler prober. Principerna är dock densamma och upplösningen beror på antalet prober samt deras fördelning. 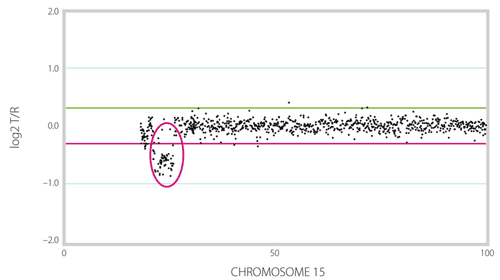 Figur 17: Ration mellan röd och grön i en punkt (en BAC klon) beräknas, och om patienten har två kopior av denna del av genomet kommer ration ligga mellan 0,8-1,2. Om patienten har en deletion kommer kloner som representerar denna region ha mindre grön inmärkning än röd, och ration blir då cirka 0,5. (Bildkälla: A Read & D Donnai, New Clinical Genetics, ©Scion Publishing Ltd) Metyleringstester De flesta gener uttrycks lika oberoende av på vilken kromosom i ett kromosompar den sitter. Det finns dock gener som bara är aktiva på den paternella eller maternella kromosomen i ett par. Detta fenomen kallas genomisk prägling (engelska imprinting) och innebär att en person måste ha en kopia av denna gen från en viss förälder. Det finns tillstånd där den normala kromosomsegregationen gått snett, eller där ett präglat kromosomavsnitt saknas. Om det är den aktiva allelen som saknas ger detta upphov till sjukdom, medan den inaktiva kromosomen kan avvaras utan problem. Genomisk prägling uppstår i samband med bildning av könsceller (spermier och ägg) genom att generna på en kromosom inaktiveras med hjälp av metylering. Genom att koppla metylgrupper på cytosin-baser i DNA kommer genen i fråga att inaktiveras. Om den andra kromosomen inte är metylerad kommer denna genkopia att användas selektivt. På samma sätt används metylering för att inaktivera den ena X-kromosomen hos kvinnor. Eftersom det finns tillstånd som orsakas av defekter i genomisk prägling finns det ett behov av att analysera metylering i enskilda gener eller kromosomavsnitt. Metyleringen kan testas med hjälp av metyleringskänsliga restriktionsenzymer. Man kan även använda PCR om man först kemiskt modifierar DNA. Sjukdomar som orsakas av defekter i genomisk prägling är t ex Prader Willis syndrom, Angelmans syndrom och Beckwith-Wiedemans syndrom. | ||||
| 8. Förvärvade mutationer | ||||
|
Verktygslådan med metoder (figur 2) ger även möjlighet att identifiera viktiga förvärvade genetiska förändringar i maligna celler. Kromosomanalys är en viktig metod inom leukemidiagnostik eftersom olika typer av leukemier ofta har specifika avvikelser, translokationer mellan kromosomer, som relativt lätt kan ses i mikroskopet. Det finns dock betydande tekniska svårigheterna med cancercytogenetik, då analyser av malign vävnad är tekniskt och biologiskt mer komplicerat. Diagnostik av kromosomavvikelser i cancerceller kompliceras bland annat av att dessa förändringar är förvärvade, så att det finns en blandning av normala celler och celler med den specifika sjukdomsframkallande förändringen i provet man analyserar. Tumörspecifika translokationer kan även detekteras med molekylära metoder. Med FISH ses en fusion mellan olika kromosomspecifika sonder och med RT-PCR kan fusionstranskript detektera när två gener smälts samman. Vidare kan samtliga metoder som används för detektion av konstitutionella mutationer också användas för att påvisa förvärvade mutationer i cancerceller. 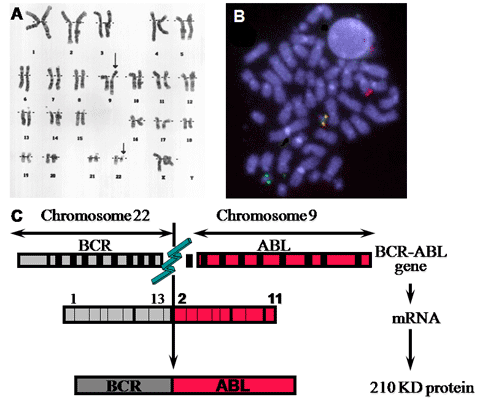 Figur 18: Bilden ilustrerar den för kronisk myeloisk leukemi karakteristiska genetiska avvikelse. A) visar en karyotyp med Philadelphiakromosomen,som en resultat av tranlokationen mellan den långa armen av kromosom 9 och den långa armen av kromosom 22. B) visar samma avvikelse med FISH sonder riktade mot ABL och BCR. C) panelen visar avvikelsen på molelylär nivå med fusionen av BCR och ABL generna som transkriberas i ett fusionsRNA och vidare translateras i ett fusionsprotein. 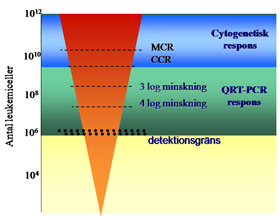 Figur 19: Modifierad efter Hughes et al. Figuren visar hur mängden leukemiceller minskar från diagnosen med behandling och känsligheten för olika metoder för remissionsbedömning, cytogenetik och kvantitativ RT-PCR (QRT-PCR). Figur 19: Modifierad efter Hughes et al. Figuren visar hur mängden leukemiceller minskar från diagnosen med behandling och känsligheten för olika metoder för remissionsbedömning, cytogenetik och kvantitativ RT-PCR (QRT-PCR).Genom att kvantifiera BCR-ABL fusionsRNAt har man ett mått på kvarvarande (antalet) tumörceller under/efter behandlingen. Metoden som används är en s.k. realtids-PCR och är baserad på amplifiering av ett specifikt DNA-fragment med hjälp av värmestabilt DNA-polymeras och korta syntetiserade DNA sekvenser; primers. Amplifieringen sker i närvaro av en tredje oligonukleotid, en s.k. hydroliseringsprobe, som är inmärkt med en flourofor i 3´ och en ”quencher” i 5´ändan och är komplementär till sekvensen mellan primrarna. Primer- och probesekvensen tillsammans med reagens och temperaturcyklingsprogram avgör specificiteten på det amplifierade fragmentet. Under PCR reaktionen hydroliseras proben av Taq DNA-polymerasets exonukleasaktivitet, och fluorescensen ökar i takt med att PCR produkt ackumuleras. Fluorescensen mäts kontinuerligt under PCR-reaktionens gång och genom att jämföra med en standard av känd kopiantal kan man beräkna kopiantal i patientprovet. Mängden av den leukemispecifika BCR-ABL RNA används som ett indirekt mått på antal leukemiceller hos patienten. Att följa mängden BCR-ABL RNA över tiden har under de senaste åren blivit etablerad som främsta uppföljningsmetod för patienter med KML. 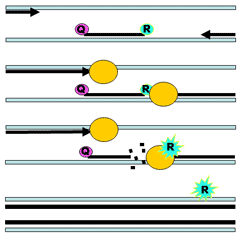 Figur 20: Figuren ilustrerar principen för realtids-PCR. PCR amplifieringen sker i närvaro av en tredje oligonukleotid, en s.k. hydroliseringsprobe, som är inmärkt med en flourofor i 3´ och en ”quencher” i 5´ändan och är komplementär till sekvensen mellan primrarna. Under PCR reaktionen hydroliseras proben av exonukleasaktivitet av Taq DNA-polymeras och fluorescensen ökar i takt med att PCR produkt akumuleras. Figur 20: Figuren ilustrerar principen för realtids-PCR. PCR amplifieringen sker i närvaro av en tredje oligonukleotid, en s.k. hydroliseringsprobe, som är inmärkt med en flourofor i 3´ och en ”quencher” i 5´ändan och är komplementär till sekvensen mellan primrarna. Under PCR reaktionen hydroliseras proben av exonukleasaktivitet av Taq DNA-polymeras och fluorescensen ökar i takt med att PCR produkt akumuleras. | ||||
| 9. Möjlighet till genetisk testning | ||||
|
Det finns idag goda tekniska möjligheter till genetisk testning av kromosomala och monogena sjukdomar. Möjligheterna kan begränsas framförallt av graden av kunskap som finns för den aktuella sjukdomen, avseende molekylär biologisk orsak, mutationsspektrum och genetisk heterogenitet. Kunskapen om de genetiska sjukdomarna ökar kontinuerligt, och därmed kommer fler och fler av dem kunna diagnostiseras med laboratorietester. Nya metoder och analyser utvecklas dessutom, vilket möjliggör förbättrad detektion av förändringar i vår arvsmassa.
| ||||
| Uppdaterad: 2007-11-28 |
Medicinsk redaktör Magnus Nordenskjöld. Kommentarer till Rula Zain.
© 2024 MedSciNet AB. All rights reserved. Legal Notices.